CRISPR-PLANT v2
A Portal of CRISPR-Cas9 Mediated Genome Editing
How to use the searching function
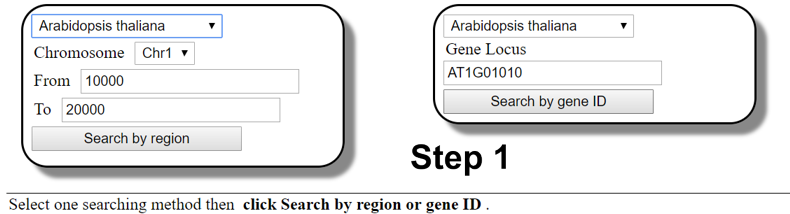
You can easily access the CRISPR-PLANT v2 data set in three simple steps
- Select plant species and chromosome region or gene of interest to start searching gRNA spacer sequences.
- Searching gRNA spacers by inputting a chromosome region;
- Searching a transcript unit or gene by inputting locus ID.
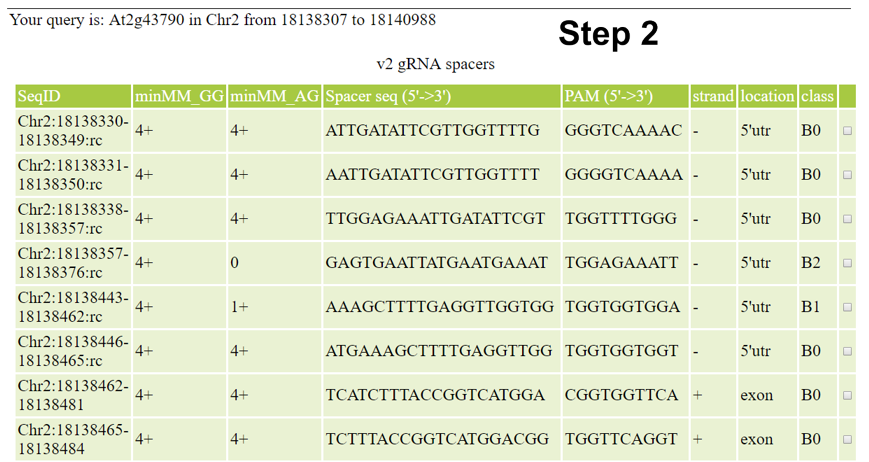
- Check the search result to identify gRNA spacer sequences that could be used to target selected gene or intergeneric region;
Currently, CRISPR-PLANT will show all Class0.0 and Class1.0 gRNA spacer sequences. We will add other gRNA spacer information in the near future. Following information will be listed for each spacer sequence:
- seqID: is populated with "Chromosome:start_position-end_position:strand". "strand" is show as empty for plus or "rc" for minus, which indicates the chromosome strand of the PAM.
- minMM_GG: minMM (minimal mismatch number) is the minimal number of differences to other NGG sites and based on the classification scheme. For example, Class 0 is populated with 4+, indicating that these spacers have 4 or more differences to other NGG spacers.
- minMM_AG: minMM (minimal mismatch number) is the minimal number of differences to NAG sites and based on the classification scheme. For example, Class 0.1 is populated with 3+, indicating that these spacers have 3 or more differences to other NGG spacers.
- seq: sequence of gRNA spacer (20 nt long). For convenience, it is shown as DNA sequence (identical to protospacer). You can use this sequence directly in your gRNA design.
- PAM: PAM with additional 7bp of the genomic sequence.
- strand: chromosome strand of the PAM.
- location: location (exon, intron, intergenic, utr, na) of gRNA targeted site. The location is based on only one splice form even some genes have alternative splicing forms. Please double check manually that the spacer is in all exons of variants you want to target.
- class: displays the specificity classificication (A0 to B2) of the spacer sequence.
- last column:a click button to start analyzing spacer sequence.
- Browse and analyze searching results to find gRNA spacers which are best for your purpose.

An analysis report will be shown after clicking the button in the last column. For restriction endonuclease (RE) cut analysis, click here to get the complete RE list used.
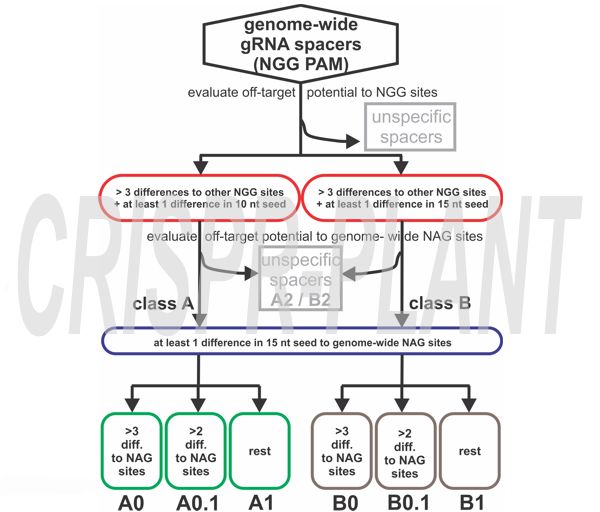
Explanation of spacer classification
- Class A: One mismatch in the 10 bp seed sequence and at least three more differences to other spacers throughout the complete spacer sequence;
- Class B: One mismatch in the 15 bp seed sequence and at least three more differences throughout the complete spacer sequence;
- Class 0: One mismatch 15 bp seed of genome-wide NAG sites and at least three more differences in addition;
- Class 0.1: One mismatch 15 bp seed of genome-wide NAG sites and at least two more differences in addition
- Class 1: One mismatch 15 bp seed of genome-wide NAG sites.
- Class 2: At least one NAG spacer sequence was found with a perfect match to the NGG spacer sequence.
We consider A0, B0, A0.1, and B0.1 gRNA spacers as highly specific specific for CRISPR-SpCas9 mediated genome editing. A1 - B2 spacers have off-target potential to NAG sites and are backup spacers if no other suitable spacer sequence was found by the user.
 If you are interesting in details about our analysis pipeline, please contact us.
If you are interesting in details about our analysis pipeline, please contact us.
Analysis strategy and bioinformatic pipeline
Previous studies identified three main types of off-targets. 1. Sequences that are similar enough to the on-target sequence to facilitate Cas9 binding and cleavage activity. 2. Sequences that have insertions or deletions when compared to the on-target sequence. This gap can occur on either the gRNA spacer or on the off-target DNA. In both cases the RNA or DNA will form a small bulge, but the remaining nucleotides will perfectly anneal in the DNA/RNA heteroduplex and facilitate Cas9 binding and cleavage. 3. sequences that do not have a 5’-NGG-3’, but a 5’-NAG-3’ protospacer adjacent motif (PAM). Even though the interaction between NAG PAMs and Cas9 are weaker when compared to NGG PAMs, the interaction is still sufficient to cause off-target cleavage if the sequences are similar enough.
We first extracted all 5’-NGG-3’ and 5’-NAG-3’ sites in a genome together with their 20 nt protospacer region. We then classified the gRNA spacer sequences adjecent to NGG sites for their off-target potential to other NGG and NAG sites. Our strategy compares each individual NGG spacer candidate with all other spacer candidates from the NGG and NAG spacer lists. In the published results, all spacer candidates provided by the pipeline have sufficient specificity to other NGG sites in the genome. These NGG spacer candidates were further divided into the two groups A and B, with A having four or more differences to other sites of which at least one difference is in the 10 nt seed region, and with B having four or more differences of which at least one is in the 15 nt seed region . We made this distinction because different studies suggest slightly different lengths for the SpCas9 seed region. Having A and B group spacers allows the user to decide for a more conservative approach with a 10 nt seed for group A. In the last step, groups A and B were further divided by their potential to NAG off-target sites based on the number of differences to these sites. In general, classes A0, B0, A0.1, and B0.1 provide sufficient differences to other NGG and NAG targets, while classes A1, B1, A2, and B2 might have potential NAG off-targets. We recommend using A1 to B2 spacers as backup only if no spacers from the high-specificity classes A0 to B0.1 can be found for the target region of interest.
Data source
| Species | Group | Genome | Download Source |
| Arabidopsis thaliana | dicot | TAIR10 | http://www.arabidopsis.org/download/ |
| Medicago truncatula | dicot | Mt4.0v1 | ftp://ftp.jcvi.org/pub/data/m_truncatula/Mt4.0 |
| Solanum lycopersicum | dicot | ITAG2.3 | ftp://ftpmips.helmholtz-muenchen.de/plants/tomato/genes/ITAG2.3_release/ |
| Glycine max | dicot | Wm82.42.v1 | https://soybase.org/GlycineBlastPages/blast_descriptions.php |
| Brachypodium distachyon | monocot | 33v1.0 | ftp://ftp.ensemblgenomes.org/pub/plants/release-33/fasta/brachypodium_distachyon/dna/ |
| Oryza sativa | monocot | RGAP Release 7 | http://rice.plantbiology.msu.edu/downloads_gad.shtml |
| Sorghum bicolor | monocot | Sorghum_bicolor_v2 | ftp://ftp.ensemblgenomes.org/pub/plants/release-32/fasta/sorghum_bicolor/ |
Working with a different genome or Cas variant? You can clone the pipeline used for analysis from our GitHub repository and customize it based on your needs!
CRISPR-PLANT is supported by Penn State and AGI.
CRISPR-PLANT©, 2018